Dockground
Model Docking Benchmark Set 1
The ability to dock protein models, as opposed to X-ray structures, is important for the progress in the docking field. This includes both specific complexes of biological interest, where the X-ray or NMR structure of one or both proteins is not available, and large-scale prediction of protein-protein interaction networks, where due to limitations of experimental approaches most structures of individual proteins have to be models. Docking applicable to protein structures of limited accuracy (models) requires approaches that are significantly different from the 'traditional' techniques designed for high-resolution X-ray structures.
To simulate the structural precision of modeled proteins, we developed earlier a procedure (Tovchigrechko et al,. Docking of protein models. Protein Sci 2002; 11:1888-1896), which distorts protein structures from PDB. The goal was to add to the database of co-crystallized protein-protein complexes the arrays of models of each subunit, with RMSD from 1 to 6 Å (the range of precision of modeled proteins). The set of distorted structures, based on the procedure, was based on now obsolete set of complexes. The procedure also did not adequately reflect structural distortions in template-based modeling.
We created a new set of model structures, with controlled structural distortions, characteristic to the real high-throughput homology models of monomeric proteins. First, we generated a dataset of homology models corresponding to the crystallographically determined monomers for 63 two-chain complexes from the DOCKGROUND X-ray benchmark set.The dataset is well populated with models at all levels of accuracy, up to 6 Å RMSD, and can provide 37% of the model benchmark structures. However, these homology models are insufficient for comprehensive benchmarking due to (a) non-uniform distribution of the models over the target pool, and (b) non-uniform distribution of the structure distortions in a particular target. For the remaining 63% of models we used the Nudget Elastic Band (NEB) method (Elber & Karplus, A method for determining reaction paths in large molecules - application to myoglobin. Chem Phys Lett 1987; 139:375-380), which generates a low-energy path between two protein conformations. For the starting point in the NEB procedure (as implemented in Amber 10) we used either homology model with the closest RMSD value below the intended one, or the initial X-ray structure (if the homology model was not available). As the end structure for the NEB trajectory, we used either the homology model with the closest RMSD value above the intended one or a structure generated by the simulated annealing, if such homology model was not available. Figure 1 shows examples of models at different levels of distortion. The schematic diagram of modeling is in the Figure 2.
Figure 1.
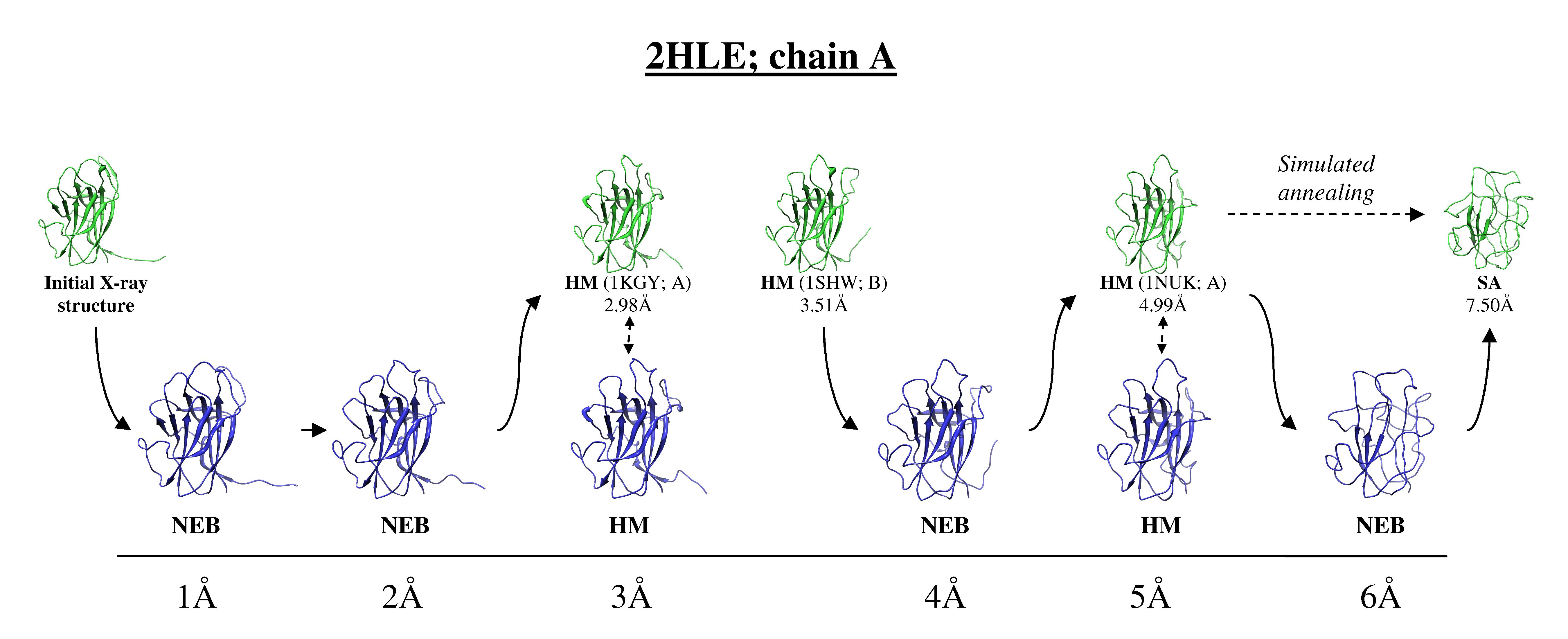
Figure 2.
Reference: Anishchenko, I., Kundrotas, P.J., Tuzikov, A.V., Vakser, I.A., 2014, Protein models: The Grand Challenge of protein docking, Proteins, 82: 278-287.
Model Docking Benchmark Set 2
The dataset contains a significantly larger number of complexes than Set 1 (165 vs 63 complexes). Thus, the benchmarking results on this set will be statistically more significant (whereas the results on Set 1 allow a direct comparison with the docking of unbound X-ray structures). The difference in the initial choice of complexes for the two sets (bound and unbound DOCKGROUND parts for Set 2 and Set 1, respectively) is the reason for small overlap between the two sets (two complexes are shared by the sets: 1oph, chains A and B, and 2a5t, chains A and B).
The built-in engine of the DOCKGROUND (Bound -> Build Database) was used to generate the initial set of the X-ray structures of binary hetero complexes with resolution 3.5 Å and better, with a well-defined interface (buried solvent accessible surface area per chain larger than 250 Å2, and at least 10 interface residues in each chain). Redundancy from the set was removed by the sequence identity 30% threshold between pairs of chains. Complexes with a protein having less than 3 secondary structure elements were excluded. We also purged complexes with monomers of substantially different sizes (where the number of residues in the larger protein is more than 3 times of that in the smaller partner). The set was visually inspected to remove complexes with coiled coil interfaces and those with interwoven chains. The cleaned set subjected to the modeling procedure contained 293 binary complexes.
The flowchart of the protocol is in Figure 3. Sequences extracted from SEQRES tag of the selected PDB files were submitted to the stand-alone I-TASSER 1.0 suite of programs (Roy et al. Nat Protoc 2010, 5:725). To ensure varying levels of model accuracy, the package was run several times with different cut-off values for the sequence identity between target and putative templates. We varied this parameter from 1 to 0.2 with 0.1 step, plus the additional value of 0.25 introduced to diversify models build at sequence identity levels close to the threshold of homology detection. Even if the native structure was selected as the top-ranked template at the threading stage, it was further subjected to the structural assembly (along with other high-ranked templates), and subsequent model refinement. The models were grouped based on their Cα RMSD to the native X-ray structure using RMSD window 0.05 Å starting from 0 Å. The structure with the lowest value of I-TASSER internal energy was selected as representative for each group. The representative models were submitted to ModRefiner program (part of the I-TASSER software suite). The Cα RMSD between full-atom models and the native structures were re-calculated and the models within the RMSD intervals 1±0.2 Å, 2±0.2 Å, … , 6±0.2 Å were selected. The procedure generated 3266 models for the initial set (92.8 % of the total 293×6×2 intended models). The final benchmark set was compiled from the complexes with both proteins having models in all six RMSD intervals (165 complexes).
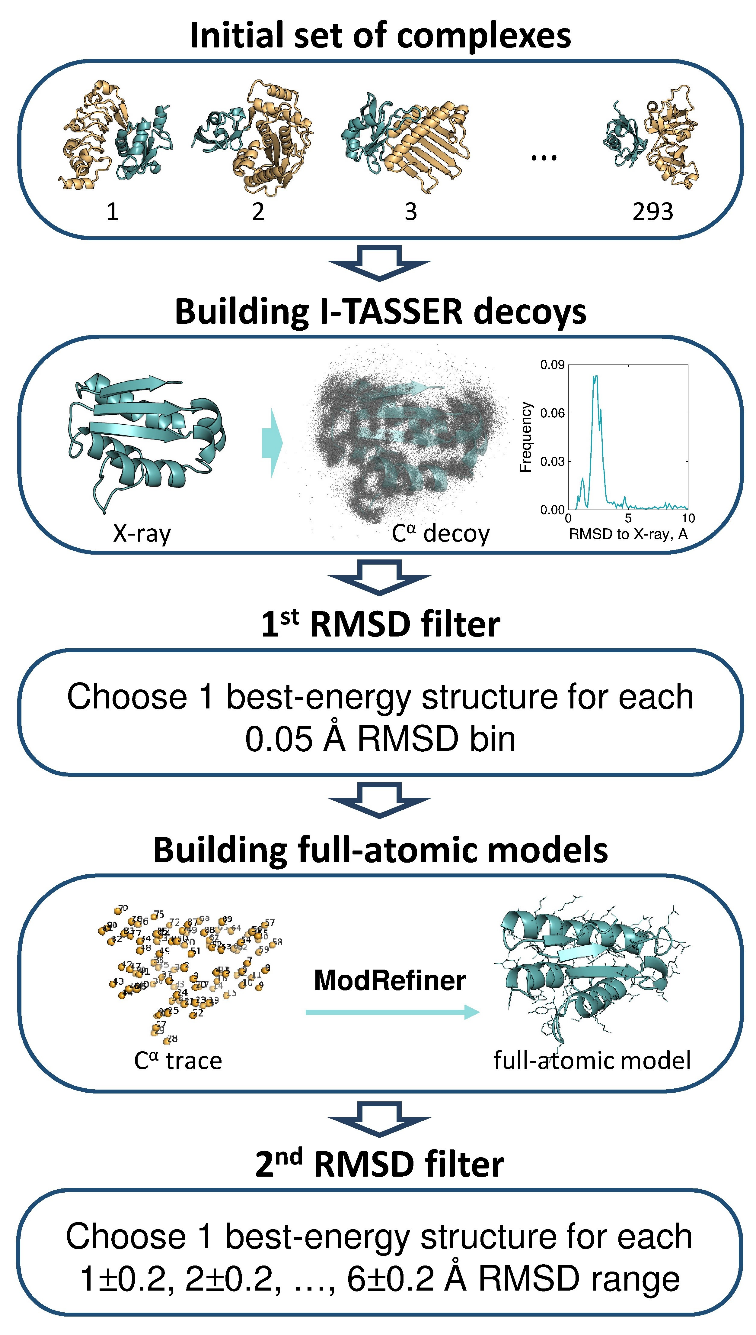
Figure 3.
Reference: Anishchenko, I., Kundrotas, P.J., Tuzikov, A.V., Vakser, I.A., 2014, Protein Models: Docking Benchmark 2, Proteins, 83: 891-897.
Model Docking Benchmark Sets Q1 and Q2
The benchmark sets of protein models for development and validation of protein docking methodologies reflect the real case modeling/docking scenario where the accuracy of the models is assessed by the modeling procedure, without reference to the native structure (which would be unknown in practical applications). The sets contain five models for each of 342 proteins (171 complexes) from the Dockground (http://dockground.compbio.ku.edu) docking benchmark set 4 (set Q1), and 1120 proteins (963 complexes) from the GWIDD (http://gwidd.compbio.ku.edu) database (set Q2). The datasets were developed by Sternberg Lab at Imperial College London and Vakser Lab at the University of Kansas within the joint GWYRE (http://www.gwyre.org) project. The models of individual proteins were generated using the batch processing facility of the Phyre2 web server (Kelly et al., Nature Protocols 2015, 10:845-858) with accuracy assessed by the Phyre ranks. The end result of the Phyre2 processing was a maximum of 20 full atom models with associated confidence scores. Conventionally, models with a confidence score < 0.9 were considered inaccurate and were excluded from the set. To avoid self-hits, a model had to have sequence identity with the template < 95%. In order to make the benchmark sets consistent and homogeneous, the final benchmark sets were compiled for the complexes with top five models of each monomer in the complex.
Reference: Singh, A., Dauzhenka, T., Kundrotas, P.J., Sternberg, M.J.E., Vakser, I.A., 2020, Application of docking methodologies to modeled proteins, Proteins, 88:1180-1188.