Dockground
Description
The database of protein-protein and protein-RNA experimentally determined structures is built from mmCIF biological unit files from PDB (Biounit files).
The chains and complexes are annotated according to their classification or other structural features. Each structure is split into pairs of interacting chains. Data is stored in a relational PostgreSQL database. The Build Database page offers the possibility to select a subset of complexes using user-defined search criteria. The database is regularly updated and annotated.
The database of protein-protein and protein-RNA complexes updates on a weekly basis and currently contains:
- 138276 PDB entries
- 763071 Chains
- 1294811 Pairwise complexes (interfaces between two Biounit chains)
Membrane Set
The database of membrane protein-protein experimentally determined structures on the basis of the Orientations of Proteins in Membranes database (OPM).
Each structure is split into pairs of chains that interact in the membrane. These chains are parsed so that only the transmembrane section of the protein is in the download file.
The database of membrane protein-protein experimentally determined complexes currently contains (as of Sept. 29th, 2021):
- 275 PDB entries
Method
Data source
Bound Set
Data in the repository are extracted from PDB CIF biounit file. In the case of multiple biounit files, the first one is used.
Membrane Set
Data in the repository are modified from OPMs the polytopic alphahelical dataset.
Selection criteria
Bound Set
- The structure's resolution must be less than 6 Angstroms
- Obsolete PDB entries are excluded
- Chains contain at least 20 residues
- RNA must contain at least 6 nucleotides
- Interface between interacting chains should exceed 250 Ų per chain.
Membrane Set
- The structure's resolution must be less than 6 Angstroms
- Chains contain at least 20 transmembrane residues
- Interface between interacting chains should exceed 250 Ų per chain.
- Transmembrane portion must contain at least one alpha helix
Redundancy Reduction
Sequence Based
Structure Based
Analysis of complexes
Bound Set
Biounit PDB entries are analyzed to extract chains that interact with each other. The MODEL tag in Biounit file has to be associated with the chain name in order to distinguish chains (Chain A in the MODEL 1 may interact with chain A in the MODEL 2). Chains are considered interacting if the interface area between them is larger than is 250 Ų (mean ASA buried by each chain). Interface areas are extracted from local files pre-computed for all PDB structures.
Membrane Set
PDB entries are analyzed to extract chains that interact with each other in the membrane.
Chains are considered interacting if the interface area between them is larger than is 250 Ų (mean ASA buried by each chain). Transmembrane segments extracted from OPM alpha helical polytopic dataset were used to extract interface areas.
These complexes were then filtred bassed on a TM score. clustering was done using highly connected subgraphs with a clustering cutoff of 0.6Å.
Additional annotations
Bound Set
- Entries are annotated with their title, resolution, experimental method, and species from mmCIF file
- If entry is a transmembrane protein
- Chains are annotated with their name, model, chain content (protein/RNA), first and last residue number, sequence, and UniProt id
- Number of domains for each chain, drawn from Pfam
- Complexes are annotated with which chains/models are involved, complex type (homo/hetero), complex content (protein/RNA), if they contain disulfide bonds ligands at their interface, mean buried ASA, number of interface residues, and number range of interface residues
Specific terms are defined below
Limitations
A major problem in compiling representative databases of protein-protein complexes is the lack of credible criteria for distinguishing complexes existing in vivo from crystal packing artifacts. The in vivo complexes have to be strong enough to be formed at the biological concentration of monomers with no help of the crystal lattice. However, the experimental data reflecting these properties are not available in many cases. In addition, the practical applicability of existing binding energy-estimating computational procedures to systematic separation of "strong" (biologically relevant) and "weak" (artifacts of crystallization) complexes is not obvious. However, functional considerations, including evolutionary factors, may provide additional help in discriminating crystal packing complexes.
Contact
If you have a general question about the database, send an email to dockground@ku.edu.
Definition of terms
The multimeric state or Oligomeric state is the number of chains that interact, at least, with one other chain in the PDB file. Thus, a dimer has a multimeric state = 2. If all interface areas are less than 250 Ų, the multimeric state is set to 0. The interface area is the sum of the mean ASA buried by interacting chain. If the PDB entry contains only one chain (e.g. interwoven chains see 2ltn, 1cov,...) then the multimeric state is also set to 0.
THUS, THE NUMBER OF CHAINS IN THE BIOUNIT FILE MAY BE HIGHER THAN THE INDICATED MULTIMERIC STATE IF SOME INTERFACE AREAS ARE LESS THAN 250 Ų (example: pdb 1gyr has a multimeric state of 2 although 3 chains are present in the PDB file: the interface area between chains B and C is 174 Ų ; example 2: 1qzv has 22 interfaces but only one A:B is greater than 250 Ų). 1qzv has a multimeric state = 2.
The Release Date refers to the initial release date of the protein structure into the PDB.
The area is the mean accessible surface area (ASA) buried by each chain in the
pairwise complex:
area = [ (ASA(chain 1) + ASA(chain 2)) - ASA(pairwise complex) ] / 2
The accessible surface area is computed by using Vorocontacts from the Voronota suite. (Olechnovič K, Venclovas Č. VoroContacts: a tool for the analysis of interatomic contacts in macromolecular structures. Bioinformatics. 2021 Dec 11;37(24):4873-4875. doi: 10.1093/bioinformatics/btab448. PMID: 34132767.).
![]()
Chain names are the names of the chains as they appear in the mmCIF file from the PDB.
Modern CIF files generally only contain one model, and label identical chains as A-1, A-2, etc. In older PDB files
this tends to correspond to chain A model 1 and chain A model 2, etc. Chain names, residue numbering, and models can differ
between PDB and CIF files for the same entry in the PDB. CIF biounit files are preferentially used in the bound databas.
![]()
The Sequence is the amino acid or nucleotide sequence of residues in the current chain retrieved from the ATOM section of the
mmCIF PDB file. The residue numbering and sequence from the ATOM section may be different from the sequence given in the SEQRES section (shown below).

![]()
![]()
![]()
![]()
![]()
The complex type specifies whether the protein complex is a homomultimeric or heteromultimeric protein complex. A homomultimeric complex is a complex where all chains have a sequence identity of at least 90% to each other, and a heteromultimeric complex is a complex containing two or more chains with sequence identities lower than 90%. This applies only to protein-protein complexes, as it's a given that protein-RNA complexes are different molecules.
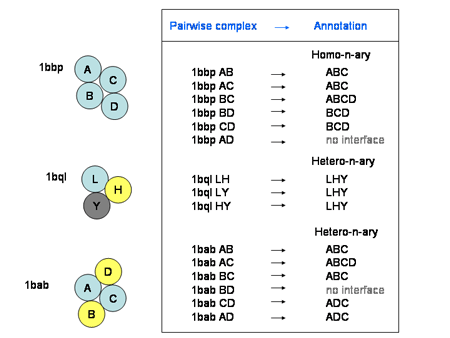
![]()
In membrane specifies if the entry is a transmembrane protein as denoted by the keywords in the PDB mmCIF file.
![]()
The complex content refers to which types of molecules are in the complex, RNA or protein (PeptideL).
![]()
The chain content refers to which type of molecule the chain consists of, RNA or protein (PeptideL).
![]()
The disulfide bonds indicates if the complex contains disulfide bonds.
The presence of a ligand at the interface (<= 5 Å from interacting residues).
Example 1GNO (ligand UOE):
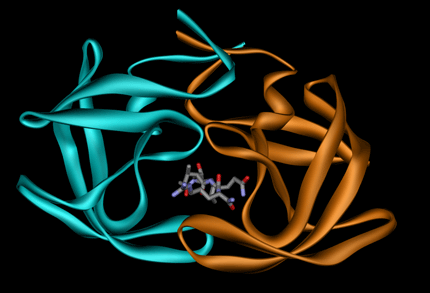
The number of interface residues field is the number of residues/nucleotides in contact with the opposite protein/RNA chain in the complex determined by changes in accessible surface area.
![]()
The interaction ranges indicates the number of the first and the last residue/nucleotide in the PDB sequence which
interacts with the other chain. Associated with the ECOD domain
boundaries, it may indicate which domain
interacts (especially when the protein contain several domains) (under development). (H. Cheng, R. D. Schaeffer, Y.
Liao, L. N. Kinch, J. Pei, S. Shi, B. H. Kim, N. V. Grishin. (2014) ECOD: An evolutionary classification of protein
domains. PLoS Comput Biol 10(12): e1003926.,
H. Cheng, Y. Liao, R. D. Schaeffer, N. V. Grishin. (2015) Manual classification strategies in the ECOD database.
Proteins 83(7): 1238-1251.)
![]()